Para fixar a posição das faces de um cristal utiliza-se um sistema de eixos de referência, conhecido como sistema de eixos cristalográficos, ele que se faz coincidir com os eixos de simetria ou com três arestas concorrente a um vértice. Estabelecido este sistema, as distâncias em que cada face corte a cada eixo medidas a partir da origem, constituem os parâmetros. Na Fig. 32 as distâncias AO (a), OB (b) e OC (c) são os parâmetros da face ABC. Se as faces cortam os eixos em seu comprimento, podem ser negativos (a ̅, b ̅, c ̅ ). Se expressam-se em forma de relação a:b:c, constituem o símbolo da face (relação axial).
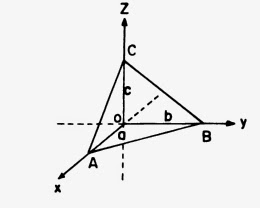
Fig. 32. Eixos cristalográficos.


Fonte:
As faces que cortam os três eixos se chamam piramidais (Fig. 33ª), as que cortam dois eixos e são paralelas a um terceiro, têm caráter prismático (Fig. 33b) e se somente cortam um eixo, são pinacoidais (Fig. 33c).
A partir de medições angulares feitas com goniômetro, e as longitudes (a, b, c) das intersecções dos planos sobre os eixos do cristal, podem calcular-se seus três eixos X, Y, Z. Para isto, o parâmetro do eixo b faz-se igual à unidade y, comparando com ele os correspondentes dos eixos a e c, se obtêm a relação paramétrica chamada relação áxica. Por exemplo, para o iodo, ortorrômbico, e o enxofre, se tem:
iodo = a:b:c = 0.661:1:1.348
enxofre = a:b:c = 0.813:1:1.904
Isto significa que se o parâmetro b tem um centímetro, o de a teria 8.1 milímetros e c terá 19 milímetros no caso do enxofre.

Fig. 33. Face piramidal (a), prismática (b) e pinacoidal (c).
ÍNDICES DE MILLER
As faces de um cristal ou os planos reticulares de uma rede cristalina podem caracterizar-se mediante um sistema de coordenadas espaciais. Miller em 1839, (citado por Azaroff3) idealizou um sistema, para descrever as faces cristalinas, que continua a ser usado até hoje em dia. Considere-se, por exemplo, um sistema de três eixos coordenados perpendiculares ente si OX, OY e OZ, como mostra na Fig. 34. Sejam a, b, c as distâncias sobre os ditos eixos que caracterizam uma célula unitária. ABC é o plano unidade da rede espacial, e a, b, c são os parâmetros. Para definir outro plano qualquer da rede, por exemplo o plano LMN, os segmentos de intersecção OL, OM e ON se expressam, primeiramente, como múltiplos ou frações dos parâmetros. Na Fig. 34 vê-se que OL = 2; OM = 3 e ON = 3 e a notação da face é 2:3:3. Os recíprocos destes números são 1/2, 1/3 e 1/3 e se encontram numa relação 3:2:2. Estes números são os chamados índices de Miller ou valores h, k e l do plano LMN, que se designa como plano (322).
O uso dos recíprocos das interseções evita o uso do conceito de infinito e as obrigatórias dificuldade de cálculo. Os recíprocos se expressam em primeira instância como frações, isto é, se dá um valor inteiro e então se referem como índices de Miller, onde h = 1/a, k = 1/b e l = 1/c, os índices de Miller são inversamente proporcionais às interações dum plano de uma rede, com os eixos eleitos.
Sempre pode-se encontrar um grupo de eixos no qual os recíprocos das intersecções são números inteiros pequenos. Para o plano ABC, os índices são (111), já que os múltiplos de a, b, c são, neste caso, iguais à unidade. Isto equivale à chamada “lei das intersecções racionais” enunciada por Haüy.

Fig. 34. Eixos cristalográficos. Idenfiticação dos planos mediante os índices de Miller.
LMN: plano paramétrico, sendo os parâmetros cristalográficos OL = a, OM = b, ON = c.
ABC: face do cristal que intercepta a/1:b/1:c/1 e cujos índices de Miller são (111)
Face FGHI intercepta a/∞:b/∞:c/1 e cujos índices de Miller são (001).
É evidente que quando uma face ou plano é paralelo a um eixo, a intersecção se encontra no infinito e o índice de Miller se transforma em 1/∞ = O.
A notação de Miller se aplica também aos planos traçados no interior de uma rede cristalina, como se observa na Fig. 35 que mostra parte de uma distribuição cúbica de átomos (Jenkins e de Vries1 7). Nesta figura se veem:
Uma série de planos em que a distância entre eles é sempre uma unidade assim:


Fig. 35. Uma série de planos com diferente índices de Miller (segundo Jenkins e de Vries1 7).
SISTEMAS CIRSTALINOS
A agrupação das formas cristalinas que apresentam os mesmo elementos de simetria permite obter-se um conjunto de formas que constitui uma classe de simetria. Por sua vez, esta associação permite agrupar os cristais de modo que em cada grupo se encontrem aqueles que possuem simetrias de mesma ordem. Tais grupos ou conjuntos constituem os sete sistemas cristalinos que agrupam 32 classes de simetria ou grupos pontuais, os quais dão origem a 230 formas cristalográficas ou grupos especiais (cf. Fig. 36).

Fonte:
As formas cristalinas provem de colocar uma base de átomos ou moléculas nas diferentes redes de Bravais e das diferentes operações de simetria e combinação delas que podem se realizar.
REDES DE BRAVAIS
Uma rede de pontos no espaço constitui uma rede de Bravais. É possível reduzir todas as formas cristalina a somente quatorze redes simples que são chamadas de redes de Bravais, ou redes
espaciais.

Fonte:

Fonte:
Se os átomos ou moléculas constituintes do sólido se situam nos vértices do poliedro, trata-se de uma rede simples; se, no entanto, contém átomos em outras posições, faces ou centro, forma-se uma rede composta. Combinando estas redes fundamentais pode-se obter quaisquer formas cristalinas.
Nas redes de faces centralizadas há uma partícula situada em cada vértice e uma no centro de cada face. Nas redes de corpos centralizados, há uma partícula em cada esquina e também, uma no centro do poliedro.
CONCEITO DE ISOMORFISMO, POLIMORFISMO E POLITIPISMO
Trata-se de modificações da composição ou estrutura de grande importância nos silicatos cristalinos.
Isomorfismo
Chama-se assim o fenômeno segundo o qual, substâncias que ainda que tenham composição química diferente, desenvolvem cristais que têm forma e estrutura similar. Diz-se que se trata de substâncias isoestruturais. Um exemplo clássico são as séries dos alúmens.


Fig. 36. Os sete sistemas cristalinos.

Fig. 37. As quatorze redes de Bravais: (1) cubica simples, (2) cúbica de corpos centralizados, (3) cúbica de faces centralizadas, (4) tetragonal simples, (5) tetragonal de corpos centralizados, (6) romboédrico simples, (7) hexagonal de faces centralizadas, (8) ortorrômbico simples, (9) ortorrômbico de bases centralizadas, (10) ortorrômbico de corpos centralizados, (11) ortorrômbico de bases e faces centralizadas, (12) monoclínico simples, (13) monoclínico de bases centralizadas, (14) triclínico simples.
O isomorfismo se apresenta com bastante frequência entre os filossilicatos, que desenvolvem séries isomorfas entre dois termos de composição diferente: montmorilonita-beidellita, beidellita-montronita, etc. Também apresentam-se séries isomorfas entre silicatos e germanatos.
Se as estruturas são as mesmas e sua composição química é similar, mas não idêntica, de modo que possam formar cristais compostos chamados soluções sólidas, as possibilidades de formar compostos isomorfos são grandes.
Nos filossilicatos, a substituição de um íon metálico estrutural por outro, por exemplo, Si por Al, Al por Fe, Al por Mg, é um fenômeno muito frequente que define muitas espécies e constitui a causa, dentre outras, de feitos tão importantes como a troca catiônica.
Polimorfismo
Chama-se assim o fenômeno segundo o qual uma substância química produz diferentes formas cristalinas. Ainda que a composição dos polimorfos sejam iguais, suas propriedades físicas são diferentes.

Fonte:



Cristobalita

Tridimita
Quando o polimorfismo se apresenta em apenas um elemento, chama-se alotropia. Exemplos são o enxofre, que pode se cristalizar no sistema rômbico ou monoclínico, e o oxigênio (O2) e o ozônio (O3) que são formas alotrópicas do mesmo elemento. Também são conhecidos os estados alotrópicos do fósforo.
Politipismo
É uma forma especial de polimorfismo. A modificação da estrutura implica em uma diferença na distribuição do empilhamento de lâminas ou placas idênticas. É um fenômeno frequente na maioria dos filossilicatos, tais como micas, caulinitas, cloritas, etc.
CRISTAIS IDEAIS E REAIS. DEFEITOS OU IMPERFEIÇÕES CRISTALINAS
Nas páginas seguintes se discutirão os diversos aspectos de simetria cristalina, concebendo os cristais como entidades perfeitas, constituídos por esferas rígidas, indeformáveis (átomos, íons ou moléculas) dispostas nos nós de uma rede tridimensional. Na realidade, geralmente isto não é assim, e se diz que os sólidos apresentam defeitos, enquanto não exibem uma ordem completa. Logo mais estes defeito serão levados em consideração, mas antes, convém definir o que se entende por cristal ideal e cristal real.
Cristal ideal é aquele no qual as fileiras de partículas que constituem a rede, permanecem sem desviar-se para todas as direções do cristal; é uma distribuição periódica que adquirem as partículas conforme a simetria de um dos 230 grupos espaciais (Azaroff³). A maior parte dos cristais naturais, equeles com que se trabalha em laboratório, desviam-se em alguma extensão dos modelos ideais e se chamam cristais reais.
Exemplo do que seria um cristal ideal

Fonte:
O termo imperfeição, ou defeito, nos sólidos, se usa para decrever qualquer desvio que sofra uma distribuição ordenada. Esta pode ocorrer por irregularidade na distribuição de suas unidades, como se sucede com as posições vazias, os átomos intersticiais, as deslocações e a substituição de uma classe de espécies por outra, como são com os átomos de impureza ou os centros F* (Sienko e Plane29). Fora os defeitos produzidos por deslocamento atômico nos cristais, é provável quem algumas imperfeições sejam produzidas em escala subatômica. Estas últimas são as chamadas imperfeições eletrônicas e são de grande importância para a explicação dos fenômenos de condutividade elétrica e outros relacionados aos sólidos (Azaroff³). Alguns autores consideram como defeito a própria superfície dos cristais, já que nela termina abruptamente a distribuição regular infinita de um cristal perfeito, deixando íons ou átomos em posições estruturais e energéticas anormais. É um fato que as superfícies neste limite têm uma influência determinante no comportamento do sistema.
(*) Considera-se que os Centros F (Farbenzentren) povenham de elétrons presos nas vacantes aniônicas, sendo o número de elétrons igual ao número de cátions em excesso nas posições catiônicas normais. São responsáveis pelas cores observadas em haletos alcalinos não estequiométricos (Sienko y Plane2 9).

Fonte:
Como consequência da entropia, é possível que a qualquer temperatura acima do zero absoluto, alguns átomos se desloquem de suas posições normais na rede a outras intersticiais, e que alguns átomos passem de suas posições normais no interior do cristal a outras na superfície. O primeiro tipo de defeito se chama defeito Frenkel, e o segundo se chama defeito Schottky.
Um defeito Schottky consiste em uma posição catiônica e outra aniônica vacante na rede, como se observa na Fig. 38. Estes defeitos se formam quando os íons deixam suas posições reticulares normais e se situam na superfície do cristal. Em um cristal simples do tipo AC, as posições vacantes implicam o mesmo número de cátions e ânions, já que a neutralidade elétrica deve se manter. Os defeitos Schottky se favorecem quando os íons são de tamanhos parecidos.


Fonte:
Fig. 38. Defeito Schottky de um cristal CA. No zero absoluto o cristal é absolutamente regular; acima desta temperatura surgem defeitos com um número igual de posições vazias tanto em ânions quanto em cátions.
No geral, a concentração de defeitos Schottky não afeta a densidade, já que é muito baixa. Todavia, há situações nas quais se chega a acumular 15% de posições vacantes, como é o caso do  -TiO, e então a diferença de densidade é mensurável (Sienko e Plane29).
-TiO, e então a diferença de densidade é mensurável (Sienko e Plane29).
-TiO, e então a diferença de densidade é mensurável (Sienko e Plane29).
O defeitos de Frenkel consiste de um íon intersticial e uma posição livre na rede. Ocorre quando alguns íons deixam sua posição normal e se situam em posições intersticiais, como se observa na Fig. 39. Para um cristal AC:

Fig. 39. Defeito de Frenkel. O cristal é absolutamente regular no zero absoluto. Acima desta temperatura se desordena; um cátion se situa em uma posição intersticial, ficando vacante uma posição catiônica equivalente.
Os defeitos de Frenkel são estimulados pela existência de cátions pequenos combinados com ânions fortemente polarizáveis.

Fonte:
b. Os defeitos Schottky e Frenkel, por ocorrer em pontos do cristal, chamam-se frequentemente de defeitos pontuais.
Outro tipo de defeito são as deslocações. Estas se produzem quando a periodicidade em uma linha de átomos em uma rede se interrompe em determinadas direções do cristal, e se chamam defeitos ou imperfeições lineares. Ocorre quando há um desalinhamento de átomos em comparação aos seus vizinhos e as deslocações mais importantes são as de aresta e as de espiral.
A deslocação de aresta consiste em uma linha reta de átomos, cada uma das quais têm um átomo de coordenação, com exceção do requerido pela estrutura do cristal. Este tipo de deslocação de indica pelo símbolo l e pode considerar-se como produzida ao inserir-se um plano da rede somente em uma porção do cristal e fechando o cristal novamente. Uma vez que o cristal se fecha, não existe nada anormal na distribuição dos átomos por este plano. A estrutura é anormal somente perto da linha ao longo da qual o plano extra termina, como pode ver-se na Fig. 40. Fora destes limites, a periodicidade é normal.

Fig. 40. Representação de uma deslocação de aresta, marcada pelo símbolo. A seta indica o plano de átomos inseridos no cristal.
Na deslocação de espiral, uma fila de átomos, cada um dos quais tem o número correto de átomos de coordenação do poliedro de coordenação está distorcido. A linha de átomos desalinhados com respeito aos vizinhos, representa um eixo no qual os planos do cristal estão deformados para produzir um efeito parecido com a espiral de um parafuso (Sienko e Plane29). Isto pode se ver em um cristal tridimensional como se observa na Fig. 41.

Fig. 41. Representação de uma deslocação de espiral. Uma parte do cristal está deslocada em comparação ao resto do cristal, terminando este deslocamento dentro do cristal.
Outro tipo de imperfeições são os chamados defeitos de plano, referidos à natureza do limite entre regiões de cristais similares ou distintos. Incluem-se também, nestes defeitos de plano, as falhas de empilhamento.
A maioria dos materiais comuns consistem no entrelaçamento de cristais muito pequenos, ou grãos, que têm uma orientação ao acaso. O limite entre dois grãos adjacentes deve então ter uma estrutura que de algum modo conforme à estrutura e orientação de ambos os grãos. Deduz-se que o limite dos grãos forma uma descontinuidade na periodicidade da rede dos cristais ou grãos e é um tipo de imperfeição de rede (Azaroff³).
Nos defeitos de empilhamento, o limite entre duas partes de um empilhamento têm sequencias de alternadas.

Fonte:
No caso dos minerais de argila, e particularmente nas argilas dos solos, os cristais são defeituosos principalmente devido às condições de sua formação, isomorfismo e politipismo. Dekeyser e Amelincko7 na Bélgica, e Hendricks1 4 nos Estados Unidos, fizeram uma série de estudos sobre o politipismo e imperfeições nos minerais de argila, especialmente em micas. No grupo das caulinitas, estes estudos se resultaram muito dificultosos devido à pequenez dos cristais e por seu alto grau de imperfeição.
Outros tipos de defeitos reticulares, incluindo fônons éxcitons, elétrons e prótons não serão considerados neste estudo.
Como assinala Greenwood¹ ³, “pode parecer surpreendente que os cristais em equilíbrio termodinâmico estejam expostos a estes defeitos, os quais surgem pela tendência espontânea de todo sistema a aumentar sua entropia ou grau de desordem. Um cristal é uma distribuição muito ordenada de íons com baixa entropia; a desorganização completa destes íons originaria um fluido com uma entropia muito maior, mas esta desorganização se opõe ao fato de que a formação de defeitos requer energia, da mesma forma que a fusão ou vaporização. A qualquer temperatura existirá uma resultante entre estas duas tendências opostas, e em equilíbrio o grau de desordem (número de defeitos) será o que seja mínima a energia de livre do cristal”.
SUBSTITUIÇÃO ISOMÓRFICA OU DIADOCICA
É pouco frequente que se apresentem minerais puros na natureza. Geralmente um ou mais íons são substituídos em certa proporção por outros íons, sendo a substituição essencialmente catiônica. Um exemplo conhecido é o da dolomita, um calcário magnésico similar à calcita (CaCO3), mas com alguns íons de Mg substituídos por íons de Ca. Tal substituição de um íon por outro em um cristal, conservando as mesmas estruturas, é o que se chama substituição isomórfica.

Calcita

Dolomita
Ao tratar das regras de Pauling23, 24, com referência à distribuição dos ânions ao redor de um cátion determinado, se viu que o número de ânions que participam é somente função da razão dos raios do cátion e do ânion (rC/rA). Dito de outro modo, a estrutura a determina primeiramente o emplaquetamento dos ânions. Por outra parte, os cátions que se associam com um grupo de ânions, o fazem em função de seu tamanho e do espaço livre que deixam os ânions (número de coordenação). Por isso, um grupo aniônico determinado será compatível com cátions de natureza distinta sempre que seus tamanhos sejam parecidos. Tais substâncias, com estruturas iguais e uma composição química similar, mas não idêntica, podem formar cristais compostos chamados soluções sólidas.
É necessário destacar que a valência dos cátions isomórficos não constitui um fator que modifique a capacidade de substituir-se uns aos outros. Deve, de qualquer modo, manter-se a neutralidade elétrica da estrutura e sempre deve compensar-se o déficit provocado pela substituição de um íon de valência maior por outro de valência menor, seja admitindo íons adicionais à estrutura, ou mudando a valência de algum íon existente. Quer dizer, deve respeitar-se as regras de Pauling.
A substituição isomórfica ocorre com muita frequência nos silicatos e outros minerais que não mostram constância na composição química e pode ser parcial ou completa. Geralmente, um íons pode substituir a outro quando seus raios iônios deferem em menos de uns 15%. Os cátions substituem a outros cátions, mas ânions como OH- e F- e outros poderiam entrar na estrutura de um mineral para substituir oxigênio.

Fonte: Do assunto Cristalografia e Cristaloquimica mandado por Profª Agna Almeida Menezes
Algumas substituições frequentes em filossilicatos são a de Mg por Ca (sempre que este tenha número de coordenação seis), Fe3+ por Al ou Fe2+ por Mg. Tais substituições não modificam o balanço elétrico e deve ser compensado pela introdução de cátions adicionais. Se o Al3+ substitui o Si4+, deve compensar-se uma unidade de valência positiva. É o caso dos feldspatos, onde alguns íons de silício estão substituídos por alumínio, e se introduz potássio ou sódio para chegar a neutralidade elétrica. Isto pode se expressar como:
Si4+ K+ + Al3+
K+ + Al3+
A razão do raio do alumínio ao do oxigênio é de cerca de 0.42, a qual é muito próxima do valor 0.414 que é o valor limite para a transição da coordenação seis a quatro. Esta é a causa de que o alumínio possa ocupar posições octaédricas e tetraédricas coordenadas por oxigênio e inclusive possa exibir ambos os tipos de coordenação é uma estrutura.
Lowenstein19 sugeriu algumas regras de regem a disposição de alumínios e silícios na coordenação tetraédrica. Se considera que quando um átomo de oxigênio é comum ou une a dois tetraedros adjacentes, somente um dos tetraedros podem estar ocupado por alumínio. Se dois íons alumínio são vizinhos a um oxigênio comum, ao menos um íon alumínio deve ter um número de coordenação maior que quatro. Esta regra explica a limitação de cerca de 50% da substituição isomórfica por alumínio, apreciável em estruturas com tetraedros polimerizados, excetuando-se as clintonitas. O incremento da carga negativa do tetraedro, derivado da substituição do silício por alumínio, deve ser compensado na estrutura por uma troca equivalente, tal como a substituição de um cátion monovalente por outro bivalente.
Geralmente o alumínio substitui o silício nos tetraedros, e o ferro, magnésio e alguns poucos cátions com tamanhos parecidos substituem o alumínio nos octaedros. Como estes íons são de tamanho maior do que os substituídos, as estruturas estão submetidas a tensões. Se o íon Al3+ (0.55 Å) substitui o íon Si4+ (0.39 Å) em uma disposição tetraédrica, posto que p Al3+ é maior que o Si4+, os átomos de oxigênio ficarão ligeiramente deslocados ao redor do alumínio, e isto significa que um tetraedro de alumínio será, necessariamente, manos estável que um de silício. Algo semelhante ocorre quando íons de Mg2+ (0.65 Å) ou Fe2+ (0.76 Å) substituem o Al3+ em uma disposição octaédrica.
Deve lembrar-se, finalmente, que as substituições isomórficas nos silicatos se produzem no momento de sua gênese. Uma vez formados, a substituição de um íons é um processo muito lento. Não deve confundir-se uma substituição isomórfica com o intercâmbio de íons, que é um fenômeno essencialmente externo.









 .
.


















 ,
,  ,
,  ou
ou  . Como pode se ver na Fig. 31, o eixo vertical é por sua vez um eixo quaternário de rotação inversão (4). A face superior P1 pode se coincidir com P1 por rotação de 90° ao redor desse eixo mais uma inversão ao redor do ponto central.
. Como pode se ver na Fig. 31, o eixo vertical é por sua vez um eixo quaternário de rotação inversão (4). A face superior P1 pode se coincidir com P1 por rotação de 90° ao redor desse eixo mais uma inversão ao redor do ponto central.
